 Hotline服務熱線:010-61006450
Hotline服務熱線:010-61006450
 簡體中文
簡體中文【全】各國匯總 | 局部外用制劑質量評價的法規要求
局部外用制劑具有可直接作用于患處發揮藥效、避免藥物的肝首過效應等優點。但由于皮膚屏障的存在,藥物通過皮膚角質層較為困難,導致起效慢,且很多藥物無法達到有效治療濃度,因此藥物釋放量及經皮滲透量的研究至關重要。本文匯總分析了國內外對局部外用制劑仿制藥的質量一致性評價要求,以指導藥物開發。
「局部外用制劑仿制藥的一致性評價要求 」
透皮制劑仿制藥一般要求與參比制劑的輔料種類及用量一致,無論FDA、EMA還是NMPA,在對處方組成的要求上大同小異,均要求輔料種類一致、用量基本相同。
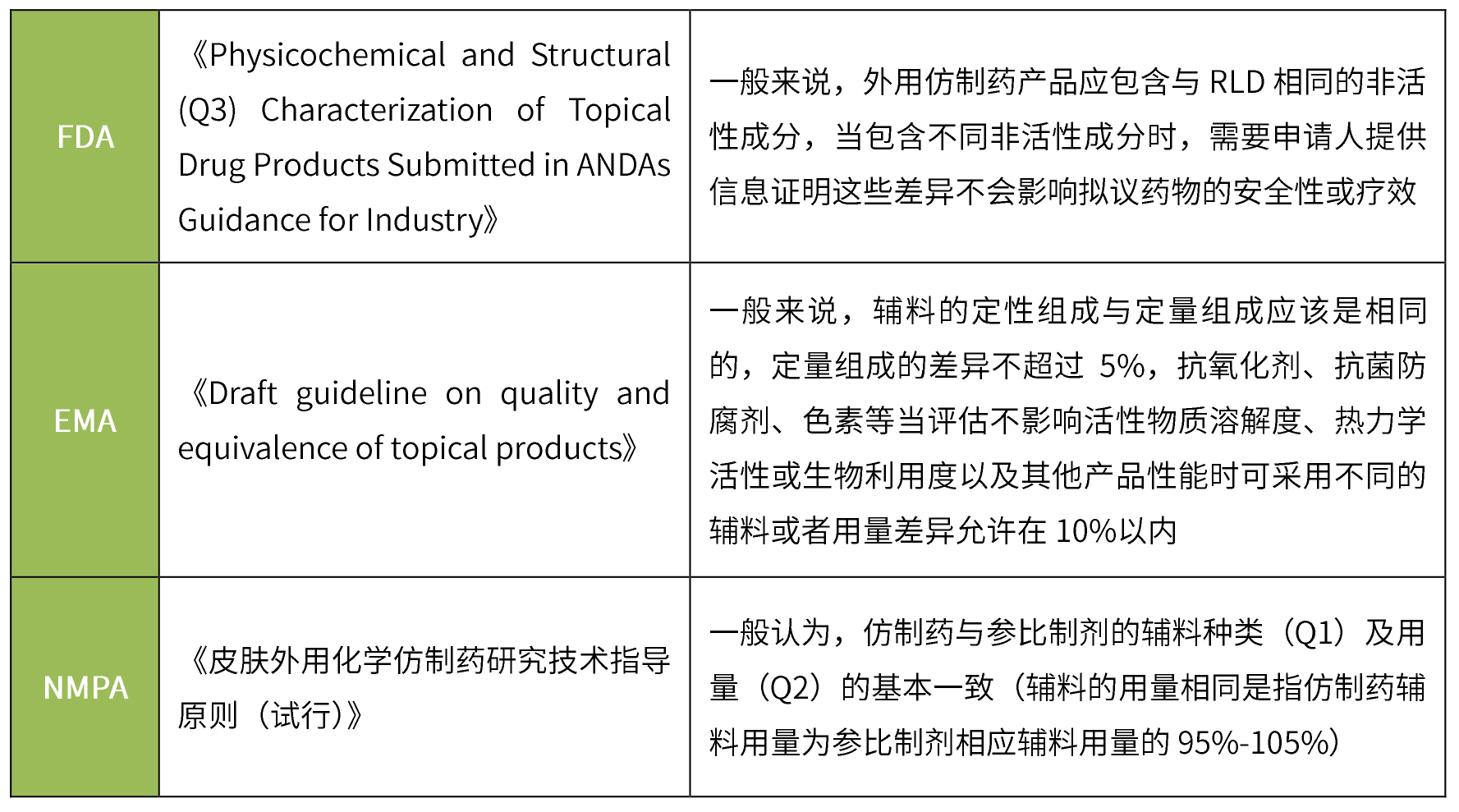
根據《皮膚外用化學仿制藥研究技術指導原則(試行)》及《新注冊分類的皮膚外用仿制藥的技術評價要求(征求意見稿)》:
-
軟膏劑、凝膠劑在開發過程中需注意對原料藥預處理工藝(如微粉化處理)、加入方式及分散手段等進行研究,以保證仿制藥與參比制劑中藥物晶型、粒度及粒度分布、含量均勻性等關鍵質量指標的一致。
-
乳膏劑、乳劑需對物料加入的順序、溶解溫度、乳化、剪切速度及混合時間等關鍵工藝參數進行研究,以保證與參比制劑的質量一致性。
3、質量控制
根據《Physicochemical and Structural (Q3) Characterization of Topical Drug Products Submitted in ANDAs Guidance for Industry》:通常建議申請人對照參比制劑對其申請的仿制藥進行Q3特性比較,并提供可接受的體外釋放試驗(IVRT)和體外滲透試驗(IVPT)的證明。其中基本Q3表征通常包括:
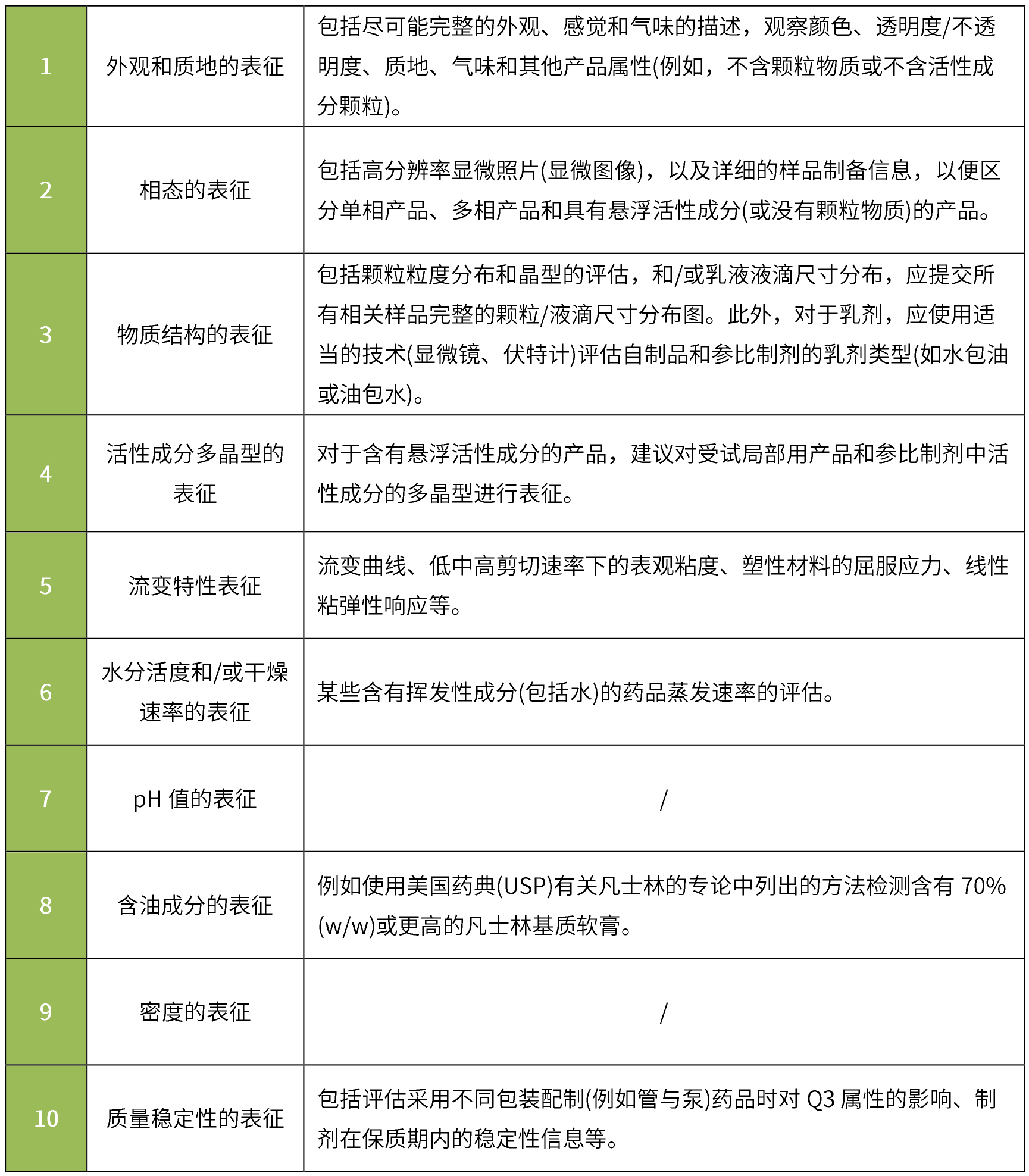
Q3相同/相似標準:在多個批次中表征的每個相關Q3屬性在參比制劑的Q3屬性表征范圍內,或在由官方機構確定的可接受變化范圍。
要求與參比制劑進行質量數據的比較,應比較劑型(單相/兩相、油包水型/水包油型等)、處方組成的定性和定量、微觀結構/物理性質(pH值、粘度、密度、表面張力、滲透壓、流變特性等)、產品性能(體外釋放試驗、藥物在皮膚和作用部位的擴散等)。
產品質量等效的對比應在能代表上市產品和生產過程的批次上進行,即達到或接近生產規模的批次。如果制造工藝和設備沒有變化,并且提供的證據表明放大不影響產品質量,則可用至少 1/10 生產規模的中試批次來表征和比較,至少應比較三個不同批次的測試產品和參比制劑,為了能夠進行統計評估,每個實驗的樣本數量至少應為每批12個單位。
對于定量質量特性,假設數據呈正態分布,測試產品和比較產品均值差異的90%置信區間應包含在參比制劑均值的+/- 10%的驗收標準內。
皮膚外用制劑的關鍵質量屬性一般包括但不限于以下項目:外觀、混懸藥物的晶型、粒度分布、液滴粒徑、流變特性、pH值、黏度、含量均勻度、微生物限度、有關物質、抑菌劑含量及抗氧劑含量、無菌(用于燒傷(除輕度 I°或 II°外)或嚴重創傷的無菌制劑)以及體外釋放試驗(IVRT)和體外透皮試驗(IVPT)等。
應對仿制品與參比制劑進行全面的質量對比研究,并提供體外釋放對比試驗和體外透皮對比試驗數據,以證明二者質量的一致性,原則上應提供多批次參比制劑的質量對比研究數據。
外用藥物在體外局部釋放的程度和速度是制劑性能的綜合體現,應提供體外釋放試驗方法的系統的研究及驗證資料,并在此基礎上對仿制藥與參比制劑進行體外釋放度對比研究。(NMPA:新注冊分類的皮膚外用仿制藥的技術評價要求(征求意見稿))。
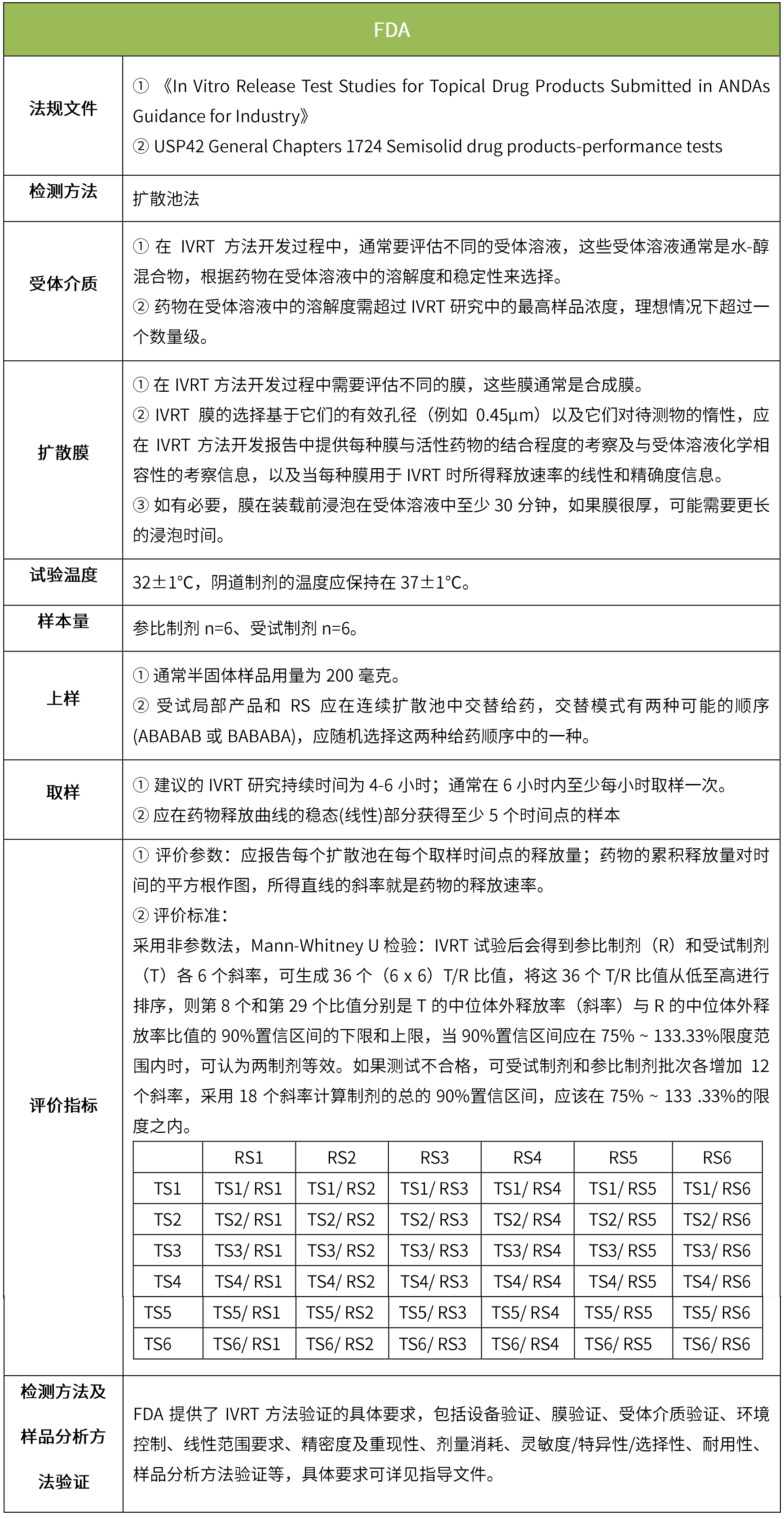
下表匯總了各國法規對IVRT的評價要求。總體而言,各國對試驗過程的要求很多都是相同的,評價標準則不同,中國的評價標準在18年征求意見稿時與FDA一致,試行版則只提及參考FDA、EMA、PMDA,歐盟和日本的評價標準與FDA也不一致。
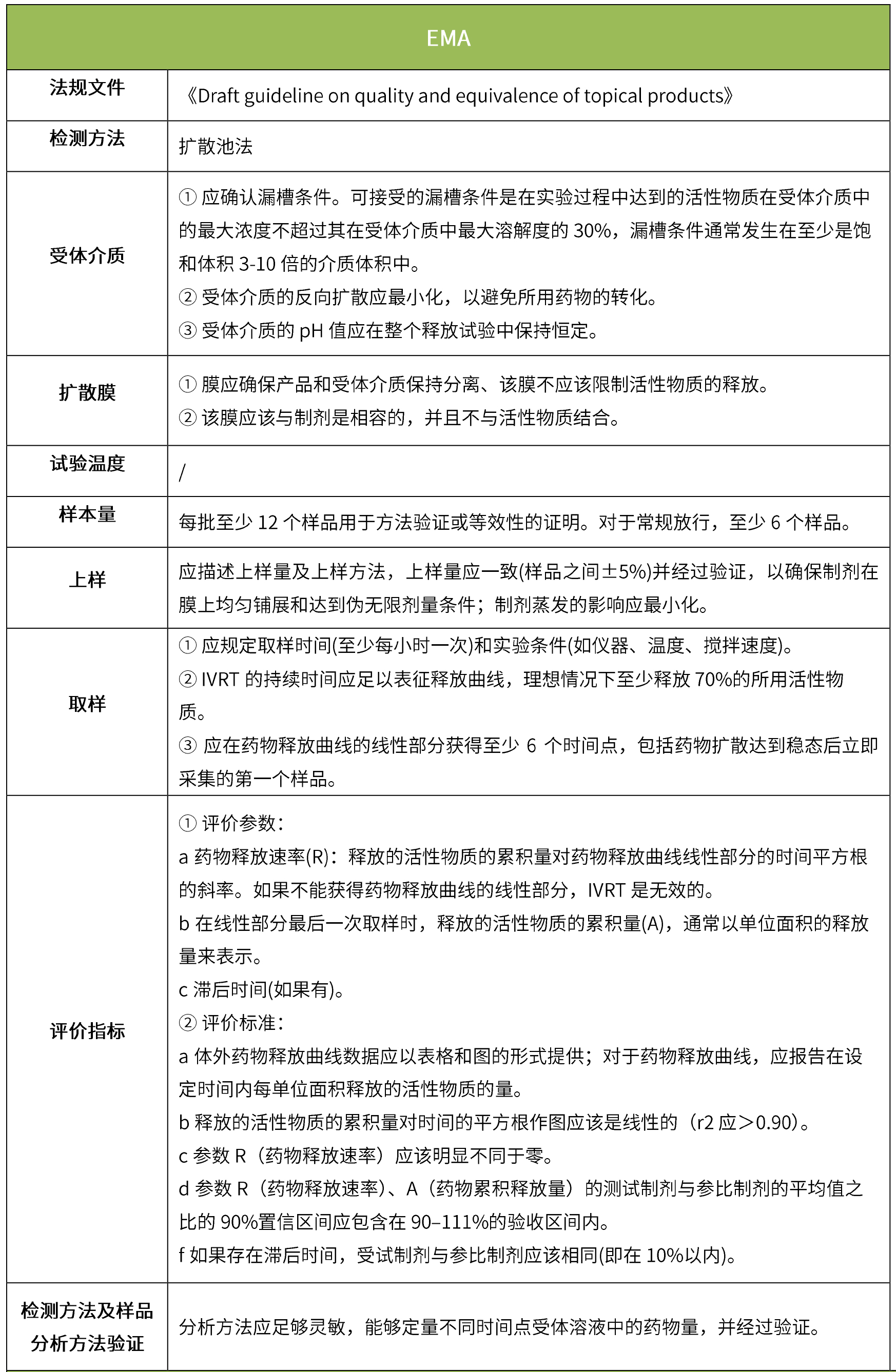
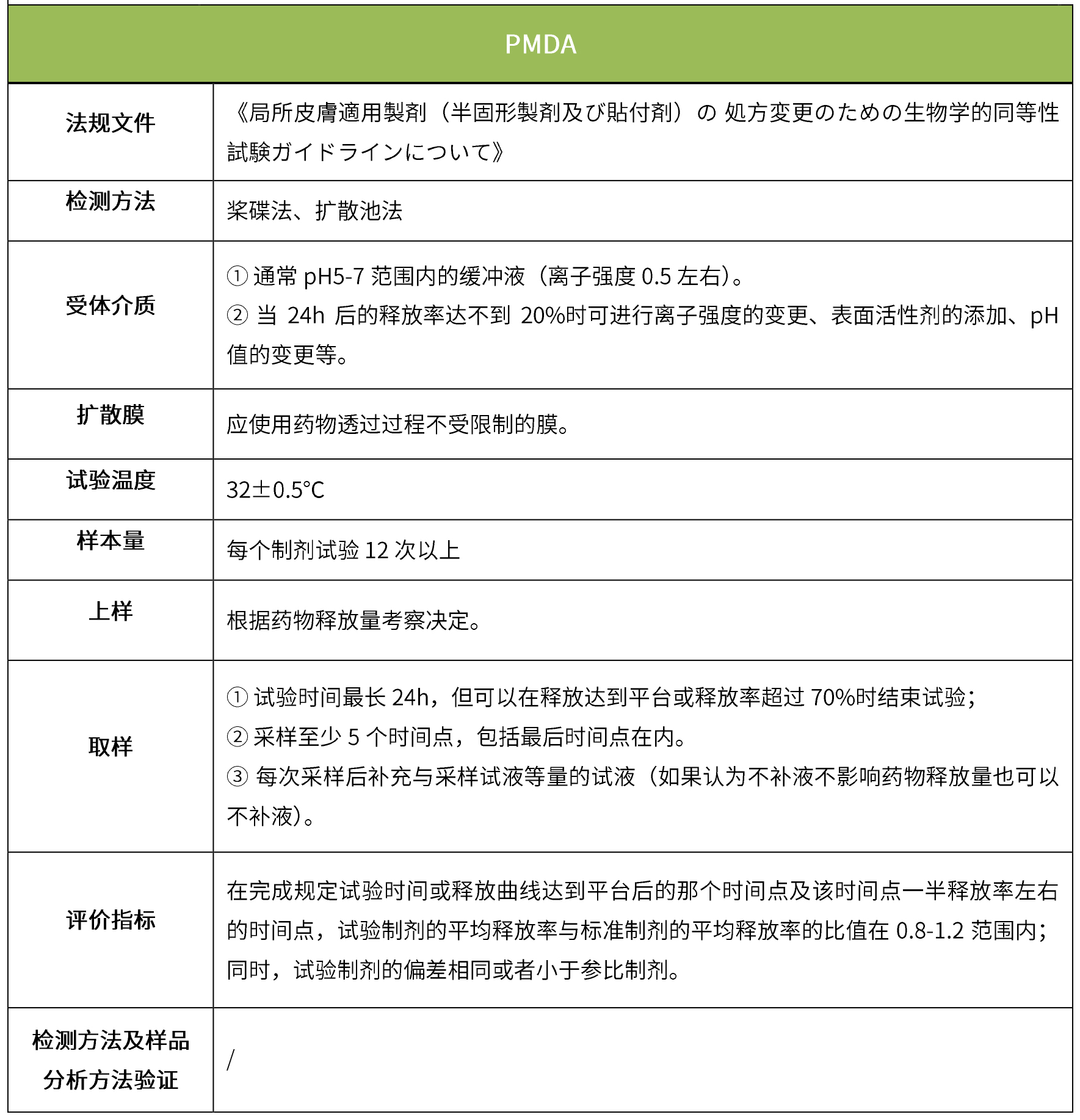
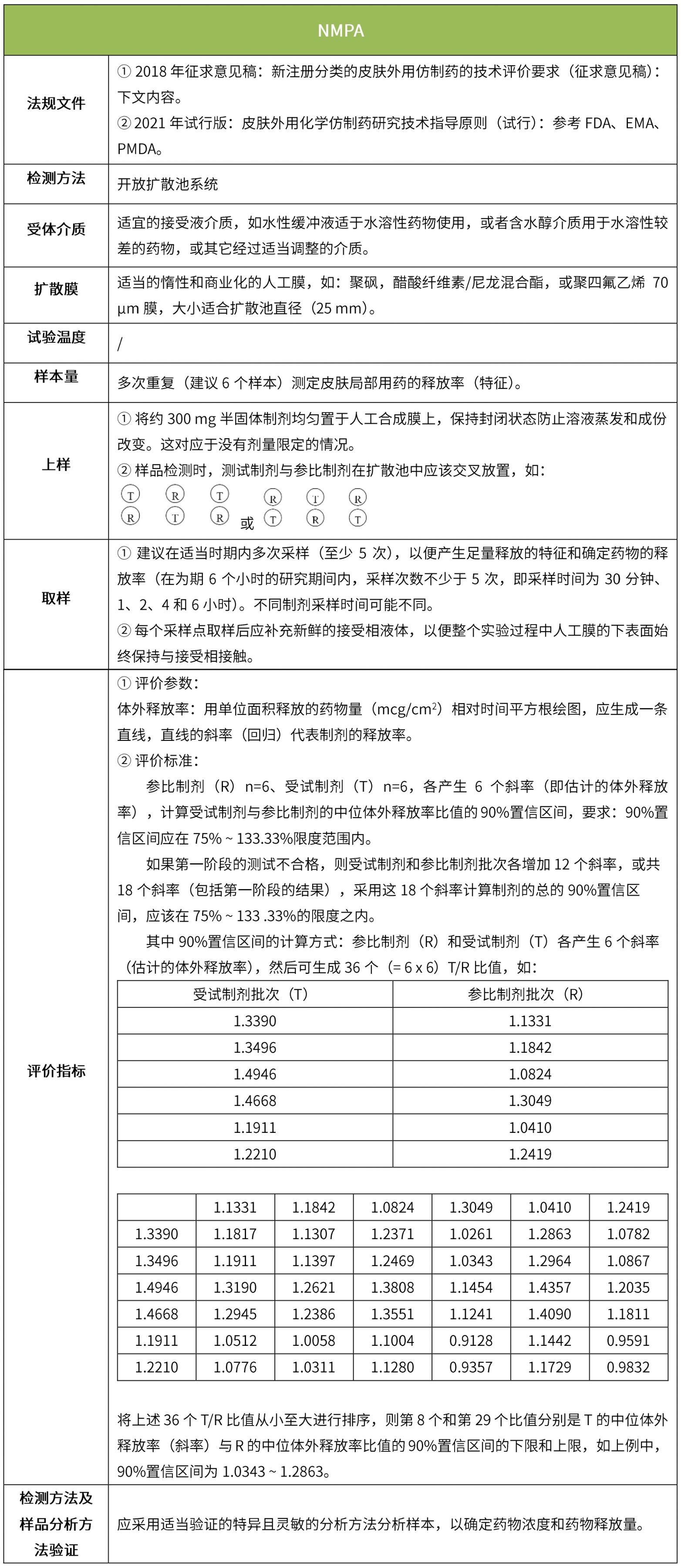
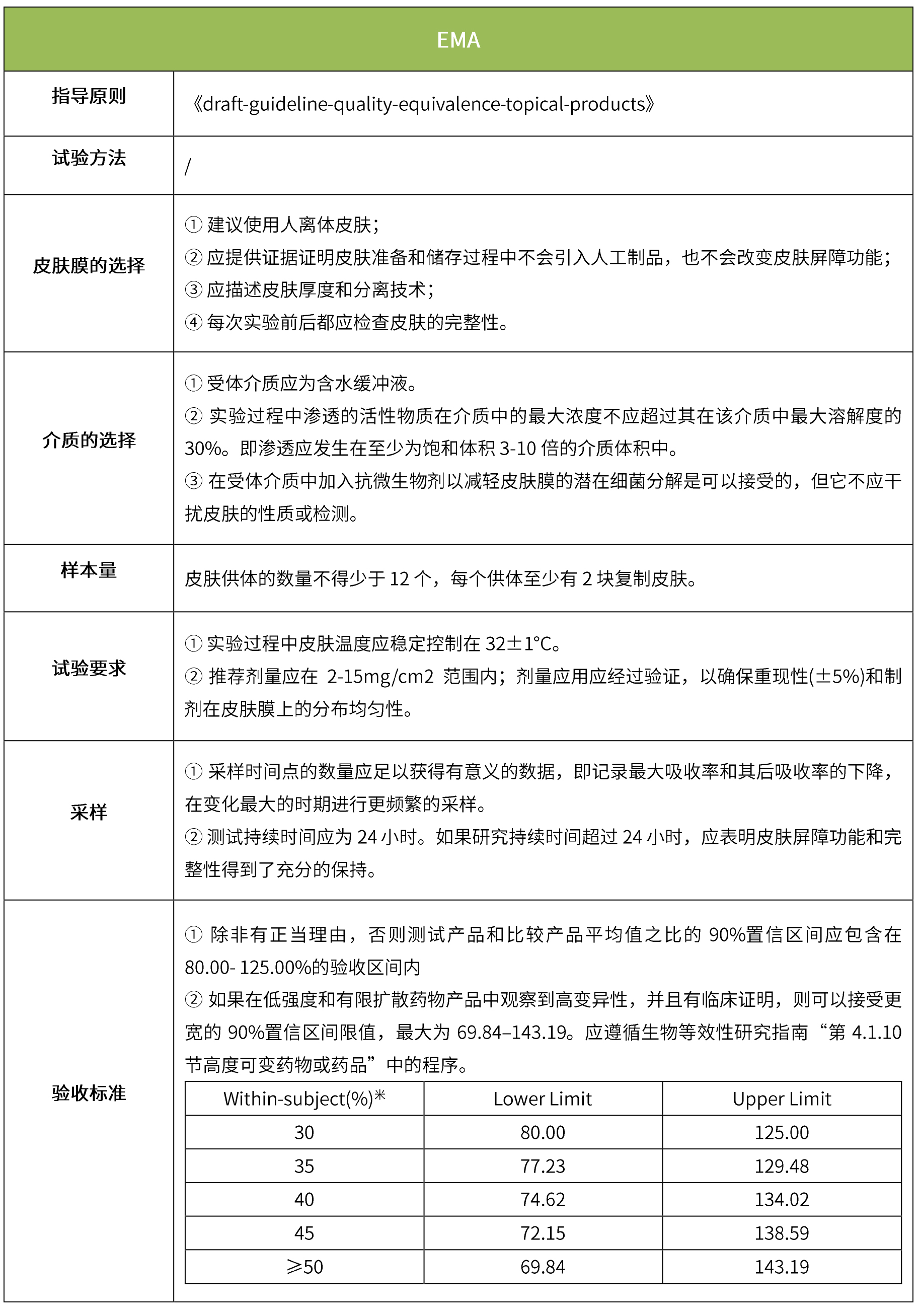
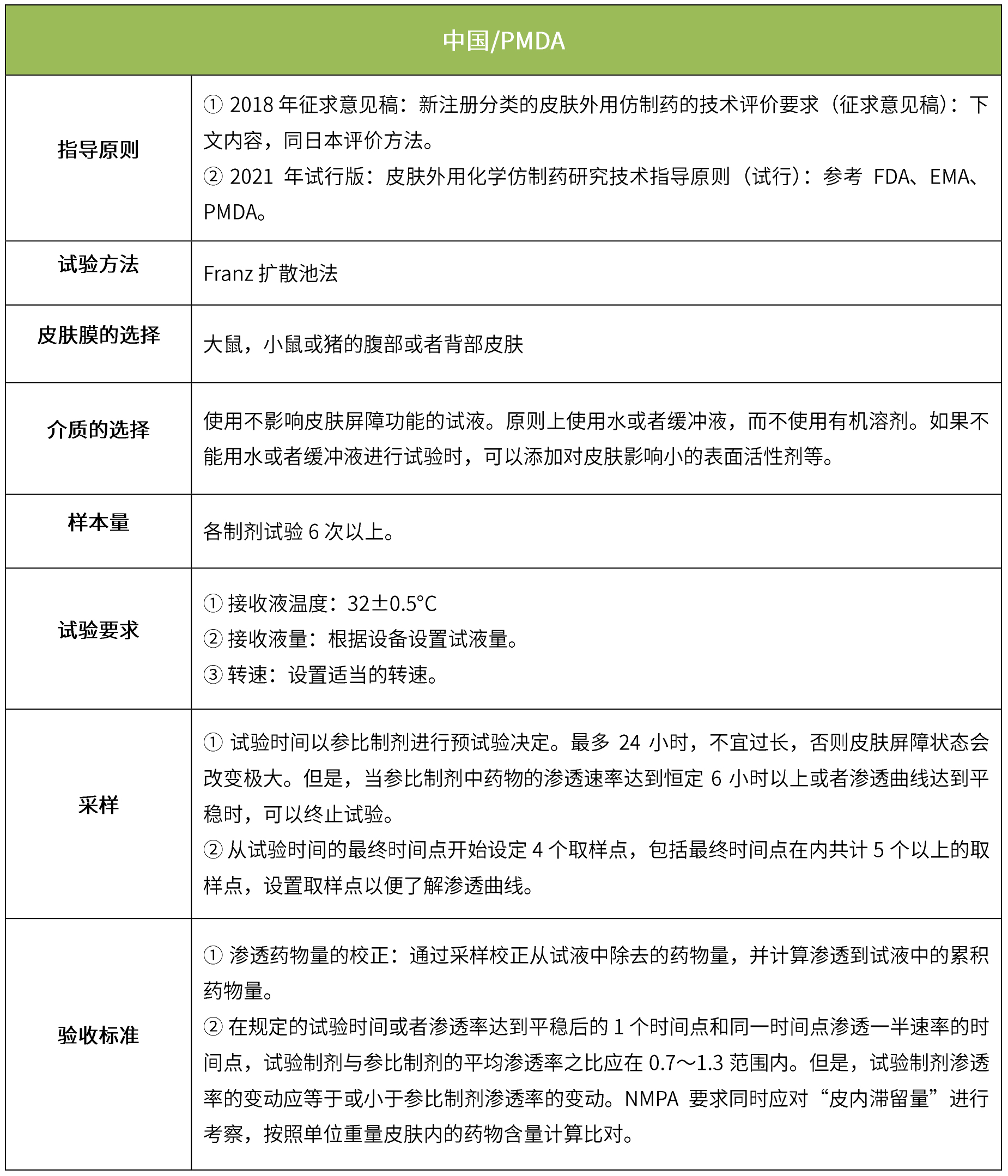
5 IVPT(體外透皮試驗)
體外透皮試驗的設計目的是模擬外用藥物在生理條件下的透皮過程,以反映外用制劑的質量;是采用特定的研究方法,動態地測量皮膚給藥后一定時間內藥物透過皮膚的速度和藥物的量,以測定藥物透過皮膚的真實生理效果。(NMPA:《皮膚外用化學仿制藥研究技術指導原則(試行)》、《新注冊分類的皮膚外用仿制藥的技術評價要求(征求意見稿)》)。
下表匯總了各國法規對IVRT的評價要求。總體而言,各國對該試驗過程的要求是不一樣的,中國的評價標準在18年征求意見稿時與日本一致,試行版則只提及參考FDA、EMA、PMDA,歐盟與FDA相比試驗過程要求上大體一致,但在樣本量和評價標準上也不一致。
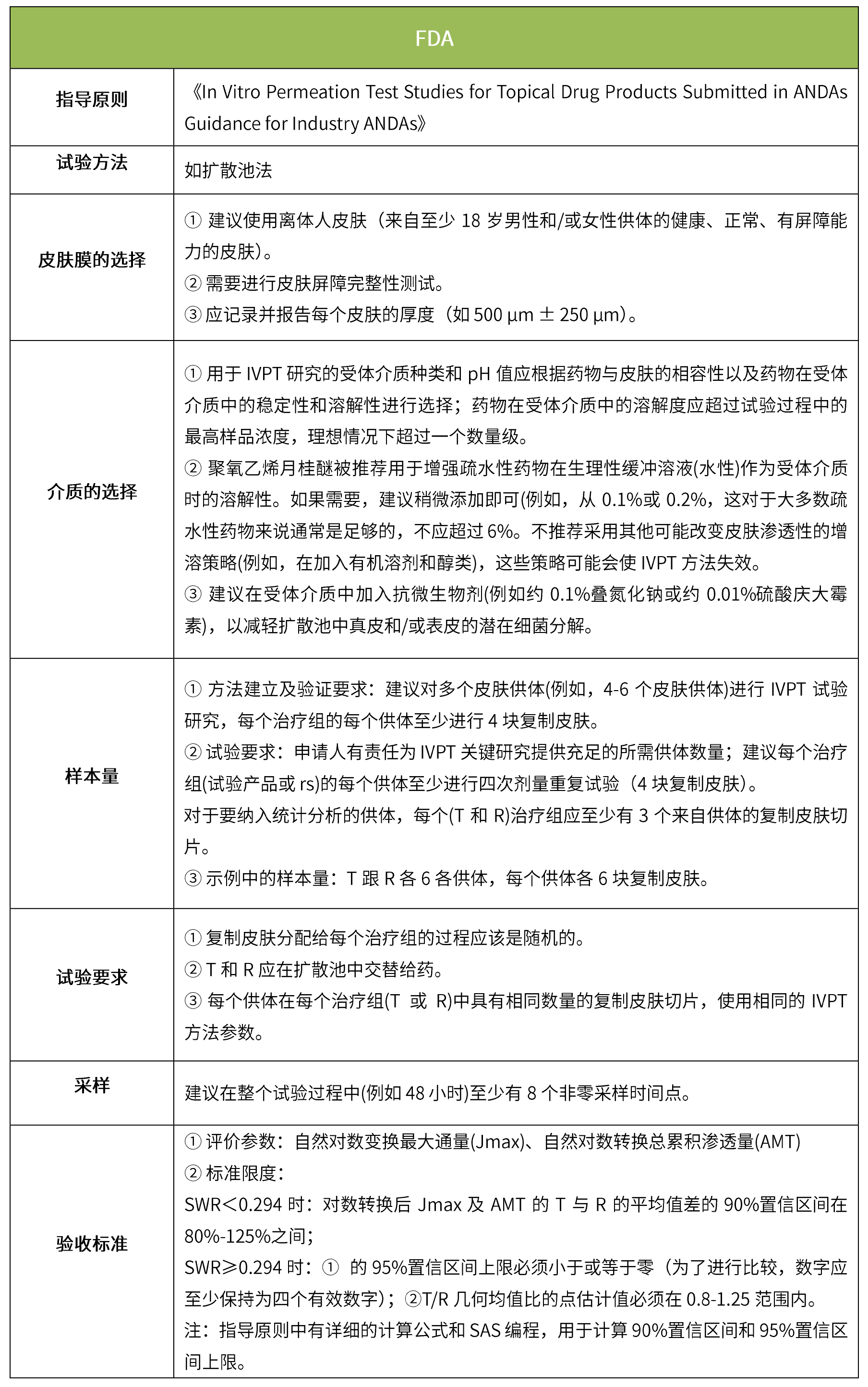
-END-


轉載聲明:未經本網或本網權利人授權,不得轉載、摘編或利用其他方式使用上述作品。已經本網或本網權利人授權使用作品的,應在授權范圍內使用,并注明“來源:新領先醫藥科技”。

 010-61006450
010-61006450 聯系地址:
聯系地址: 技術市場部:
技術市場部: 010-61006450
010-61006450
